| Em 1955 Jerome Conn(1) descreveu o caso de uma paciente de 34 anos que apresentava quadro clínico de hipertensăo severa e hipopotassemia, revertido inteiramente após a remoçăo cirúrgica de um adenoma da supra-renal direita que secretava aldosterona. A nova síndrome clínica – síndrome de Conn ou hiperaldosteronismo primário (HAP) – caracterizava-se por aumento da excreçăo urinária de potássio, taxas elevadas de secreçăo de aldosterona e diminuiçăo da atividade plasmática de renina, além de hipopotassemia e hipertensăo arterial. Descriçőes subseqüentes de casos de HAP mostraram que năo só adenomas solitários, mas também a hiperplasia bilateral das supra-renais, causavam as mesmas alteraçőes clínicas e laboratoriais. Outros subtipos de HAP, como formas familiares, hiperplasia adrenal primária e carcinoma adrenocortical, foram publicados entre a década de 1970 e o início dos anos 1990(2-4). Em que pese o melhor conhecimento da síndrome de Conn e suas causas, até 1990 a prevalęncia de HAP na populaçăo de hipertensos ficava entre 0,05% e 2%(5, 6). Era entăo considerada causa incomum de hipertensăo arterial. |
Nos últimos anos, vários grupos independentes de investigadores comprovaram que a doença năo só era muito mais freqüente, como ocorria em hipertensos normopotassęmicos( 7, 9). Contribuem para este cenário os critérios de rastreamento mais eficientes e o desenvolvimento de métodos ancorados nos mais recentes avanços da bioquímica, da genética e da biologia molecular. Novas técnicas de imagem, como a tomografia computadorizada, também tiveram um papel importante.
Atualmente admite-se que uma estratégia de diagnóstico corretamente aplicada ŕ populaçăo de hipertensos poderá identificar pelo menos um entre dez hipertensos atendidos em clínicas de hipertensăo como portadores de HAP(10).
A mesma proporçăo pode se repetir em hipertensos que procuram serviços de atençăo médica primária(11). Embora năo haja unanimidade, entre os grupos que estudam HAP, quanto ŕ maior prevalęncia da síndrome, năo se pode negar que o tema tem grande importância, principalmente se levarmos em conta que um número razoável de pacientes năo está se beneficiando de tratamentos mais específicos e até mesmo curativo de sua hipertensăo arterial.
Causas de hiperaldosteronismo
Adenomas secretores de aldosterona e hiperplasia adrenal
bilateral ou hiperaldosteronismo idiopático săo as causas mais
freqüentes de HAP(7). Entre os adenomas secretores existe
uma variante, o adenoma secretor responsivo ŕ angiotensina,
em que a secreçăo de aldosterona năo é autônoma, respondendo
ao ortostatismo e ŕ angiotensina(12). Outra variante
é a hiperplasia adrenal unilateral ou hiperplasia adrenal
primária, que tem características bioquímicas idęnticas ŕs
de adenomas secretores, apresenta níveis urinários elevados,
de 18-OH-cortisol e 18-oxocortisol, podendo ser controlada
pela adrenalectomia unilateral(13). A ocorręncia familiar
de HAP é rara, menos de 3% dos casos de HAP, existindo
um subtipo herdado por transmissăo autossômica dominante,
o FH-I, que se associa a um gene híbrido. O FH-I
apresenta-se clinicamente com hipertensăo severa e resistente
aos agentes anti-hipertensivos usuais; os pacientes săo
habitualmente normopotassęmicos e o quadro clínico pode
ser revertido pela administraçăo de dexametasona(14). O outro
tipo familiar de HAP, o FH-II, năo responde ŕ supressăo
com glicocorticóide, năo se relaciona ŕ presença de mutaçăo
genética e se manifesta por características clínicas e bioquímicas
indistinguíveis dos adenomas solitários ou da hiperplasia
adrenal bilateral(14). Os carcinomas adrenocorticais
săo muito raros e acabam por ser descobertos como massas
tumorais funcionantes, geralmente volumosas, com mais
de 6cm de diâmetro.
primário
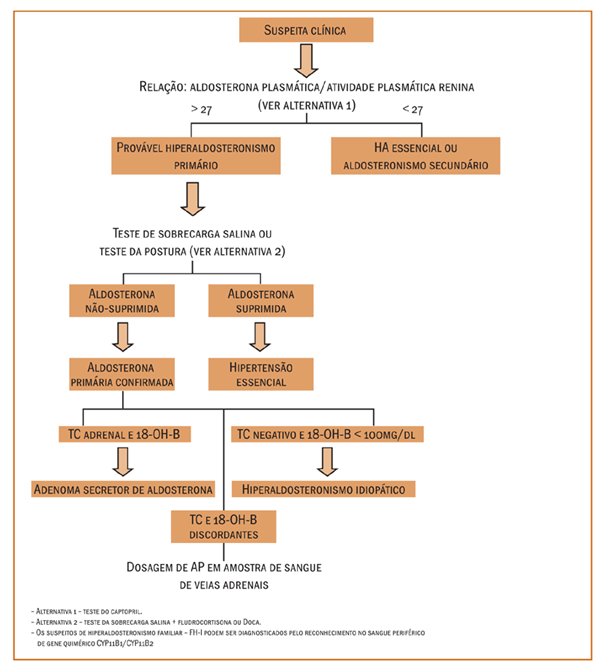
Diagnóstico de hiperaldosteronismo
primário (ver Figura 1)
Testes de screening
Embora a hipopotassemia esteja presente em muitos
pacientes com HAP, é um indicador pouco sensível e pouco
específico da doença. Por exemplo, a administraçăo de dieta
hipossódica prolongada interfere nos mecanismos de perda
de potássio pelo rim, diminuindo sua excreçăo urinária, o
que atenua a hipopotassemia. Por outro lado, algumas séries
de casos tęm mostrado que apenas 20% a 30% dos pacientes
com HAP săo portadores de hipopotassemia (14-15).
Níveis séricos de potássio
Atividade plasmática de renina (APR)
Por conta da expansăo do volume extracelular, a
APR está suprimida em quase todos os pacientes com
HAP (< 1ng/ml/h), e năo aumenta apropriadamente
(> 2ng/ml/h) após 120 minutos em posiçăo ereta, restriçăo
de sódio ou administraçăo de furosemida(16). O teste
é um indicador mais sensível que a hipopotassemia ou os
níveis elevados de aldosterona no plasma ou na urina.
Contudo tem pouca especificidade, pois a APR diminui
com a idade. Além disso, pacientes que apresentam níveis
baixos de APR e aldosterona normal tęm sido rotulados
como hipertensos essenciais com renina baixa(17),
embora a maioria dos mesmos năo tenha sido cuidadosamente
estudada para se excluir HAP normopotassęmico.
Relaçăo aldosterona plasmática/atividade
A relaçăo AP/APR vem sendo considerada um dos testes
de screening mais confiáveis para o possível diagnóstico
de HAP, mostrando altas sensibilidade e especificidade(4).
Uma relaçăo (quando a APR for expressa em ng/ml/h e a
AP, em ng/dl) maior que 25 é sugestiva, e quando igual ou
maior que 50, virtualmente diagnostica HAP. Numerosos
investigadores, de várias partes do mundo - Lim (Reino
Unido)(10), Gordon (Austrália)(7), Young (Estados Unidos)(16)
e Loh (Cingapura)(18) -, e que vęm publicando as maiores
séries de casos de HAP, mostram que a relaçăo AP/APR
aumentou em mais de dez vezes a freqüęncia de detecçăo da
doença, năo só em hipertensos hipopotassęmicos, mas também
naqueles com hipertensăo resistente e normopotassęmicos
(Tabela). Para um destes autores, a relaçăo AP/APR
teve sensibilidade de 93% quando comparada ao teste de
supressăo com fludrocortisona ou ŕ presença de HAP comprovado
cirurgicamente(10). Por outro lado, em estudos
prospectivos envolvendo pacientes com AP/APR elevada, o
tratamento com espirolactona diminuiu significativamente
a pressăo arterial e o número de medicamentos necessários
para controlar a hipertensăo(19).
plasmática de renina (AP/APR)
Embora os valores da relaçăo AP/APR, a partir dos quais
o diagnóstico de HAP passa a ser sugestivo, năo sejam os
mesmos nas várias séries de casos publicados, já que foram
diferentes as condiçőes locais e as características dos grupos
estudados(20, 21), os seguintes pontos devem ser observados
para o melhor rendimento do teste: 1) drogas antihipertensivas
como inibidores de ECA, betabloqueadores e
clonidina deverăo ser suspensas duas a quatro semanas antes
da obtençăo das amostras de sangue; quanto ŕ
espirolactona, este prazo aumenta para seis a oito semanas.
Quando for necessário, controlar a hipertensăo com bloqueadores
1 pós-sinápticos, diuréticos tiazídicos e bloqueadores
de canal de cálcio; 2) os pacientes precisam estar
repletados de potássio, já que a hipopotassemia reduz os
níveis de aldosterona; 3) as amostras de sangue deverăo ser
obtidas no meio da manhă e duas horas após, em posiçăo
ereta devido ŕ variaçăo circadiana e aos efeitos da postura
sobre os níveis de AP e APR; 4) atentar se a AP está expressa
em unidades internacionais (pmol/l) e se o laboratório utiliza
a renina plasmática ativa ao invés da APR em ng/ml/h.
Neste caso, a relaçăo AP/renina plasmática ativa estará acima
de 140 no HAP e abaixo de 100 em hipertensos essenciais.
Entre 100 e 140, recomenda-se a repetiçăo do teste(5).
Teste do captopril
Nos pacientes com produçăo autônoma de aldosterona,
a administraçăo de captopril tem pouco ou nenhum
efeito sobre as concentraçőes de aldosterona e níveis de APR,
podendo, assim, diferenciar casos de HAP dos pacientes
hipertensos essenciais. Em hipertensos essenciais, com sistema
renina-angiotensina normal, o captopril diminui a angiotensina
II e a aldosterona, embora níveis elevados de renina
estejam presentes devido ŕ inibiçăo da angiotensina. O protocolo
é muito simples e consiste na mensuraçăo dos níveis
plasmáticos de aldosterona antes e 2 horas após a administraçăo
por via oral de 25mg de captopril. Recomenda-se
que os pacientes permaneçam em jejum por 12h, que o
sangue seja coletado na posiçăo sentada e que anti-hipertensivos
como espirolactona, bloqueadores betadrenérgicos e
clonidina sejam suspensos(5). O teste tem correlaçăo altamente
significativa com os valores obtidos após sobrecarga
salina(22), é bem tolerado, a pressăo arterial permanece estável
durante o estudo e parece năo ser afetado pelas variaçőes
individuais da ingesta de sódio. A reduçăo em mais de 20%
dos níveis de aldosterona pós-captopril - usualmente menos
que 410pmol/l (< 15ng/dl) - é considerada resposta
normal. Este teste tem sensibilidade excelente, entre 90% e
100%, mas sua especificidade é de 50% a 80%(4).
Testes confirmatórios
A relaçăo AP/APR năo estabelece o diagnóstico de HAP.
A síndrome precisa ser confirmada pela demonstraçăo de
que a secreçăo de aldosterona é autônoma. Entre os protocolos
freqüentemente utilizados, e sobre os quais se tem maior
experięncia, podemos citar os que se seguem.
Teste da sobrecarga salina
O teste de sobrecarga salina consiste em se administrar
por via oral, durante tręs dias, aproximadamente 10g de
cloreto de sódio ou, como alternativa, dois litros de soro
fisiológico, durante quatro horas, visando a suprimir a produçăo
de aldosterona que ocorre com a expansăo do volume
extravascular. Se a excreçăo urinária de 24h de aldosterona,
após sobrecarga oral de sódio, for superior a 28mmol/
dia a 39mmol/dia (10mg/dia a 14mg/dia), na presença de
excreçăo urinária de sódio maior de 250mmol/dia, o diagnóstico
de HAP está praticamente confirmado, com excelentes
sensibilidade e especificidade (96% e 93%, respectivamente)(
8). Se utilizarmos os níveis fisiológicos de AP após
expansăo do volume intravascular com soro, valores acima
de 280pmol/d (> 10ng/dl) também săo fortemente consistentes
com o diagnóstico de HAP(23).
Alguns autores combinam a sobrecarga salina por via
oral com a administraçăo de acetato fludrocortisona, 0,1mg
a cada seis horas(4). Níveis de AP superiores a 240pmol/l
(8,5ng/dl) ou de aldosterona urinária maiores que 12mg/
24h confirmam que a secreçăo de aldosterona năo foi suprimida,
o que praticamente confirma o diagnóstico da HAP.
Pode-se usar a desoxicorticosterona (Doca) em substituiçăo
ŕ fludrocortisona, administrando-se 10mg do mineralocorticóide
de 12h/12h, por via intramuscular, durante tręs dias
consecutivos, e medindo-se a excreçăo urinária de aldosterona
24 horas antes do início e no terceiro dia após o uso de
Doca. Em hipertensos essenciais pode haver diminuiçăo de
pelo menos 50% na aldosteronúria, enquanto nos pacientes
com HAP a queda é inferior a 10%. Atentar para o fato
de que pacientes com hiperplasia adrenal bilateral comportam-
se, em relaçăo ŕ supressăo com Doca ou fludrocortisona,
como hipertensos essenciais. Assim, a năo-supressăo da
secreçăo de aldosterona, além de caracterizar o HAP, permite
o diagnóstico diferencial entre adenoma secretor de aldosterona
(ASA) e hiperplasia adrenal bilateral (HAI). É
útil também a relaçăo da AP (em ng/dl) e as concentraçőes
de cortisol no plasma (em mg/dl), que costuma ser inferior
a 3 nos pacientes com HAI. Em contrapartida, em portadores
de ASA, o valor é geralmente superior a 3, mostrando
a autonomia do adenoma.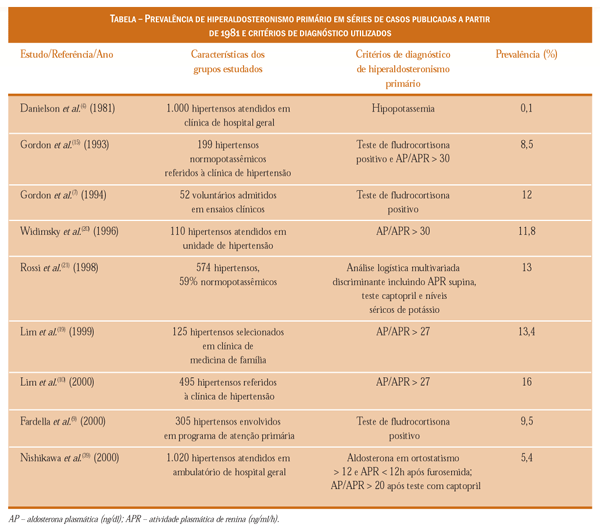
Deterioraçăo da funçăo diastólica ventricular esquerda
e aumento do intervalo QT tęm sido descritos com o protocolo
de sobrecarga salina associado ŕ fludrocortisona, principalmente
se o paciente tiver hipertensăo arterial ou insuficięncia
cardíaca subclínica(24).
Teste da postura
Outro protocolo que permite distinguir os ASA da
HAI é a resposta da AP e do cortisol ŕ mudança
postural(25). Em indivíduos normais e hipertensos essenciais,
as concentraçőes plasmáticas de aldosterona se elevam
pelo menos 50% duas a quatro horas após
deambulaçăo ou adoçăo da posiçăo ereta. A acurácia do
teste aumenta quando se medem simultaneamente os
níveis de cortisol e AP. Em pacientes com ASA, ambas as
variáveis năo se elevam com a mudança postural, podendo
mostrar até reduçăo quando em ortostatismo. Em
contrapartida, os portadores de HAI, quando assumem
a postura ereta, respondem ŕ mesma com aumento de
dois a tręs níveis da AP e da APR em relaçăo aos níveis
basais(26). O valor preditivo do teste da postura em distinguir
os ASA e casos de HAI é de 90%, embora sua
especificidade seja menor devido aos subtipos de ASA e
de HAI. O declínio postural da AP também acontece
nos subtipos (FH-I), que săo controlados pela administraçăo
de dexametasona.
A dosagem na urina de 24h dos compostos cortisólicos
híbridos 18-oxigenados-18-oxocortisol e 18-OH-cortisol
pode diagnosticar a variante FH-I e diferenciar os ASA dos
casos de HAI. Ambos săo importantes marcadores de FHI,
podendo apresentar níveis dez vezes superiores aos valores
de referęncia. Estes níveis estăo discretamente aumentados
no ASA e normais em pacientes com HAI(27).
Por outro lado, os níveis sangüíneos de 18-
hidroxicorticosterona (18-(OH)-B), que é produto intermediário
da via de síntese da aldosterona, estăo usualmente
acima de 2.800nmol/ml (100mg/dl) em indivíduos com
ASA, diferenciando-os, assim, daqueles com HAI que mostram
níveis significativamente menores(28) (Figura).
Diagnóstico radiológico do
hiperaldosteronismo primário
A tomografia computadorizada (TC) é o método radiológico
mais utilizado para a avaliaçăo anatômica das adrenais
em pacientes suspeitos de HAP. Os ASA, quando tęm entre
1cm e 3cm de diâmetro, podem ser detectados pela TC,
principalmente se o exame for feito utilizando-se cortes de
3mm.
Tendo-se em vista os resultados de várias séries de casos
de ASA publicados recentemente(8, 9), chega-se ŕ conclusăo
de que a TC năo pode ser considerada padrăo-ouro de diagnóstico.
Por exemplo, a TC detectou apenas 47% dos ASA
comprovados cirurgicamente em uma das maiores séries de
casos de HAP, năo diagnosticando um quarto dos tumores
medindo menos de 1cm de diâmetro(14). Isto é significativo
quando se sabe que um entre cinco adenomas de suprarenal
tem menos de 1cm de diâmetro. Nesta mesma série, a
TC mostrou-se claramente equivocada, diagnosticando
massas nas adrenais contralaterais, mas năo nas ipsilaterais.
Em outra série menor, estudada por McAlister e
Lewanczuk(29), dos 18 pacientes com HAP, 13 tiveram o
diagnóstico de ASA comprovado cirurgicamente e os demais
mostraram HAI năo-funcionante. É importante ressaltar
que 2% a 10% dos tumores diagnosticados pela TC
năo săo produtores de aldosterona (14). Mas em pacientes
com HAI as adrenais poderăo parecer normais ou bilateralmente
aumentadas, com ou sem micronódulos(30).
Os dados mencionados acima, outras evidęncias da prática
clínica e estudos de séries de casos tęm apontado que a
coleta de sangue por cateterismo seletivo de uma das veias
adrenais é o único método confiável para diferenciar o aumento
unilateral de produçăo de aldosterona, passível portanto
de correçăo cirúrgica, de casos de HAI. Deve-se, também,
considerar que os adenomas responsivos ŕ angiotensina
II săo bioquimicamente indistintos dos casos de HAI. Isto
reforça, ainda mais, a importância do exame como método
indispensável para este diagnóstico. O cortisol plasmático
deve ser estimado simultaneamente, mas em alíquota de
sangue coletada separadamente, para que se comprove a
origem da amostra obtida. O gradiente cortisol/veia adrenal
e cortisol/veia cava inferior deve ser igual ou superior a 3
para que os resultados possam ser considerados confiáveis.
Em măos experientes, e quando o cateterismo da veia adrenal
direita for feito com sucesso, o procedimento faz o diagnóstico
em mais de 95% dos casos. Contudo a incidęncia de
insucesso pode chegar a 25%(4).
Mutaçőes genéticas e
Em 1992, Lifton et al.(31) descreveram um teste utilizando
metodologia Southern blot para reconhecer no sangue
periférico o DNA de um gene híbrido que firmaria o
diagnóstico de HAP familiar, subtipo FH-I. Tręs anos depois,
Jonsson et al.(32) relataram um método mais rápido
utilizando PCR, com os mesmos objetivos. Estes foram uns
dos passos mais importantes para a caracterizaçăo de formas
primárias e familiares de aldosteronismo, passíveis de serem
controláveis pelo uso de dexametasona administrada em
doses orais de 0,5mg a 2mg por dia(33). Estes casos, até entăo,
eram diagnosticados pela demonstraçăo de supressăo
da secreçăo de aldosterona pelo teste da dexametasona sujeito
a falsos positivos e falsos negativos(9, 34). A partir daí
inúmeros casos de HAP foram reconhecidos em famílias de
várias partes do mundo(9, 35, 36), estabelecendo-se que o subtipo
da doença é causado pela herança de um gene mutante ou
quimérico formado pela combinaçăo do CYP11B1, gene
que codifica a enzima 11ß-hidroxilase (promove a 11ßhidroxilaçăo
da DOC, para formar corticosterona na zona
glomerular e zona fasciculata, e do 11-deoxicortisol, para
formar cortisol na zona fasciculata) e o gene CYP11B2, que
codifica a aldosterona sintase (participa da conversăo na zona
glomerulosa da cortiscosterona em aldosterona) e que, nos
casos de HF-I, sintetiza 18-OH-cortisol e 18-oxocortisol.
O gene mutante resulta na expressăo ectópica de aldosterona
sintase, que passa a ser controlada pelo ACTH, e năo
pela angiotensina II (31, 37, 38) Embora raros, menos de 3%
dos pacientes com HAP, os portadores de FH-I podem ser
reconhecidos numa fase bastante precoce, pré-clínica, de
HAP quando nem hipertensăo arterial nem hipocalemia
estăo presentes. Como já foi mencionado, o outro subtipo
de HAP familiar, FH-II, năo se associa a mutaçăo de gene
híbrido, tem características clínicas, bioquímicas e
morfológicas indistinguíveis de casos de ASA ou HAI e parecem
ser mais freqüentes que o subtipo FH-I(34).
hiperaldosteronismo primário